
在单细胞rna测序中,细胞数量过多会导致双细胞比例的增加,但实际情况往往是其他指标表现不佳。例如:
Estimated Number of Cells:估计检测到的高质量细胞数Fraction Reads in Cells:高质量细胞的序列数百分比Mean Reads per Cell:每个高质量细胞的平均序列数Median Genes per Cell:每个高质量细胞的基因数中值Total Genes Detected:所有细胞检测到的基因总数Median UMI Counts per Cell:每个高质量细胞的平均UMI数
尽管如此,许多研究者仍然倾向于测序更多的细胞,约1万个细胞左右通常是可以接受的。但如果实验环节出现问题,测序2万个或更多的单细胞可能会带来麻烦。
在《Single-Cell RNA Sequencing of Peripheral Blood Mononuclear Cells From Pediatric Coeliac Disease Patients Suggests Potential Pre-Seroconversion Markers》这篇文章中,单个样品测序了近2万个单细胞,总共分析了19,663个单细胞。然而,通过严格的质量控制步骤,过滤后剩余的细胞数不到1万个,这是一个不错的结果。具体的过滤参数并不严苛,主要是确保每个细胞至少检测到200个基因,这是单细胞转录组数据处理中的常规设置(min.features = 200)。以下是相关代码:
library(Seurat)# https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz## Load the PBMC datasetpbmc.data
通过这样的过滤,可以看到作者的策略是有效的。下图展示了过滤前后的效果:
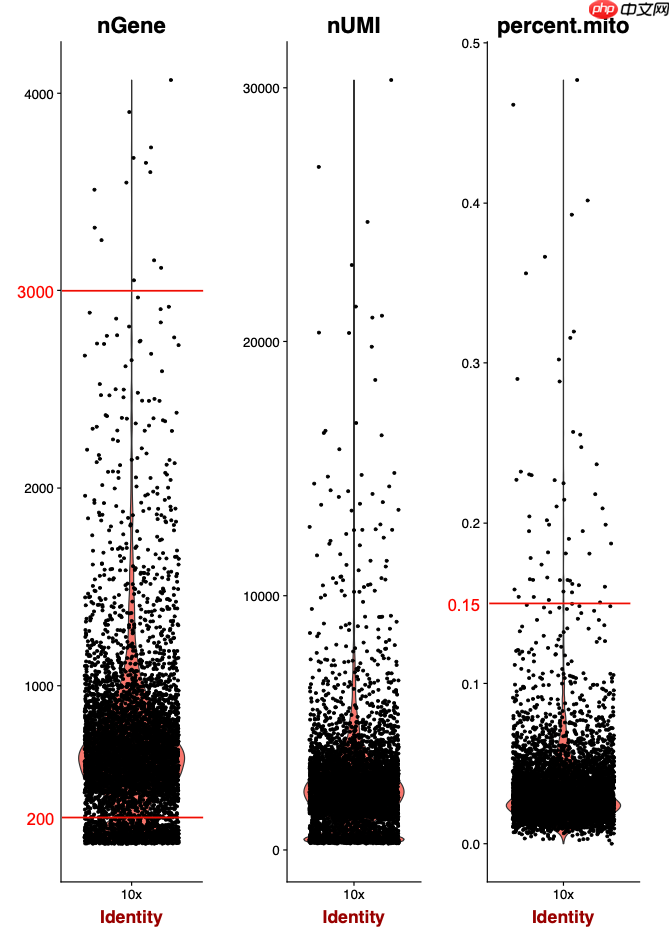
对于这篇文章的附件中提供的表达量矩阵,尽管质量略有瑕疵,但进行降维聚类分群和生物学命名时问题不大:
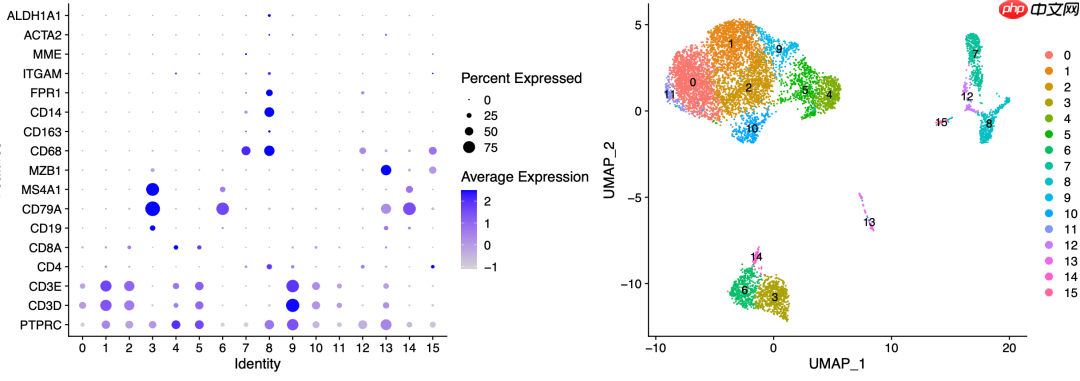
从降维聚类分群图中可以清楚地看到不同免疫细胞的分群。以下是用于定义细胞亚群的代码:
#定义细胞亚群celltype[celltype$ClusterID %in% c(7,8,12,15),2]='Myeloids'celltype[celltype$ClusterID %in% c(0,1,2,9,10,11),2]='CD4'celltype[celltype$ClusterID %in% c(4,5),2]='CD8'celltype[celltype$ClusterID %in% c(3,6,14),2]='Bcells'celltype[celltype$ClusterID %in% c(13),2]='plasma'
大部分文章都会进一步细分免疫细胞亚群,包括淋巴系(T、B、NK细胞)和髓系(单核、树突、巨噬、粒细胞)两大类。文章通常选择进行大量的差异分析和注释来讲述故事。基础的单细胞转录组数据处理步骤包括:
上游分析流程课题多少个样品,测序数据量如何过滤不合格细胞和基因(数据质控很重要)过滤线粒体核糖体基因去除细胞效应和基因效应单细胞转录组数据的降维聚类分群单细胞转录组数据处理之细胞亚群注释把拿到的亚群进行更细致的分群单细胞转录组数据处理之细胞亚群比例比较
最基础的步骤往往是降维聚类分群,参考前面的例子,可以帮助大家掌握单细胞聚类分群注解的基本方法。
以上就是单个样品测序了近2万个单细胞怎么办的详细内容,更多请关注创想鸟其它相关文章!
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。
如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 chuangxiangniao@163.com 举报,一经查实,本站将立刻删除。
发布者:程序猿,转转请注明出处:https://www.chuangxiangniao.com/p/27453.html
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 


























